What is Phylogenetics?
Phylogenetics is the study of the evolutionary relationships between species. [1] Phylogenetic trees are a visual representation of species' relationships. [2] Phylogenies can be constructed using any biological feature, including gene and protein sequences, behavior, or morphology. [1] While phylogenetic trees are only hypotheses predicting evolutionary relationships, they are useful tools to help explore gene evolution and biological similarity.
How to Make a Phylogenetic Tree
Sequence Formatting
Before creating a phylogenetic tree, the DNA, RNA, or amino acid sequences must be gathered for the gene or protein of interest as well as its homologs in each of the additional species that one desires to be placed on the tree. These sequences must be formatted into a FASTA file format for use with phylogeny-construction software.
Sequence Alignment
Once the relevant FASTA sequences have been gathered, they must be aligned based on structural similarity. This alignment can be performed by programs such as Clustal Omega or Mega.
Tree Generation
Using the newly-aligned sequences, phylogenetic trees can be constructed to display the predicted evolutionary relationships between the species of interest. Slightly-different results can be expected based on the method of generation used, as each method uses different statistical techniques to determine its tree's structure. Two of the more common methods for phylogenetic tree construction are the maximum likelihood and neighbor-joining methods.
Before creating a phylogenetic tree, the DNA, RNA, or amino acid sequences must be gathered for the gene or protein of interest as well as its homologs in each of the additional species that one desires to be placed on the tree. These sequences must be formatted into a FASTA file format for use with phylogeny-construction software.
Sequence Alignment
Once the relevant FASTA sequences have been gathered, they must be aligned based on structural similarity. This alignment can be performed by programs such as Clustal Omega or Mega.
Tree Generation
Using the newly-aligned sequences, phylogenetic trees can be constructed to display the predicted evolutionary relationships between the species of interest. Slightly-different results can be expected based on the method of generation used, as each method uses different statistical techniques to determine its tree's structure. Two of the more common methods for phylogenetic tree construction are the maximum likelihood and neighbor-joining methods.
- For the Maximum Likelihood method, a phylogenetic tree is built according to another generation method, but following generation, the tree's branches are modified to maximize the likelihood that the tree's branches and nodes are correctly placed. [3]Maximum likelihood is the more comprehensive tree generation method.
- The Neighbor-Joining method begins as a star-shaped tree lacking any branching points. The most similar species are then linked together as 'neighbors'. [4] The algorithm continues linking 'neighbors' in this fashion until all nodes are linked together. [4] Of the two methods, neighbor-joining is faster and less computationally-demanding at the cost of accuracy.
GLI3 Phylogenetic Trees
Maximum-Likelihood Model
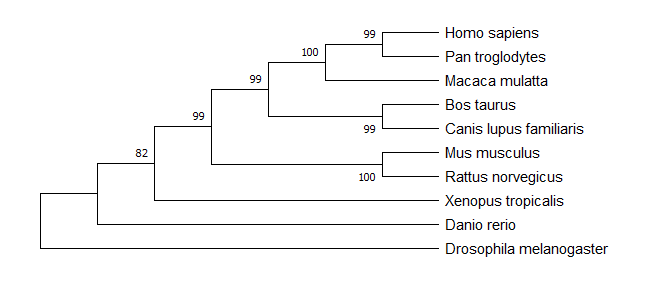
Neighbor-Joining Model

Conclusion
Both the maximum-likelihood and neighbor-joining models produced identical trees predicting the evolutionary history of GLI3. The trees demonstrate how all of the species displayed evolved their GLI3 homologs from a common ancestor, and that the species with more similar embryogenesis processes have more recently-diverged GLI3 homologs. The trees group together mammals, as expected, as they are the most genetically-similar species of those listed on the tree. If gene conservation is a concern when selecting a model organism, these phylogenies demonstrate that mus musculus has a GLI3 homolog that has diverged from the human GLI3 fairly recently (on a genetic timeframe) and is likely to be highly-conserved.
References
- Berkeley University. (n.d.) The family tree. Retrieved from https://evolution.berkeley.edu/evolibrary/article/0_0_0/evo_04
- EMBL-EBI. (n.d.) Phylogenetics: An introduction. Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
- NCBI. (July 2004). Maximum Likelihood. Retrieved from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
- Yang Z, Rannala B. (March 2012). Molecular phylogenetics: principles and practice. Retrieved from https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
Images:
Header: http://www.evogeneao.com/en/learn/tree-of-life
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
